
Sean Hormozian [1], Dayken Dawson [2], Megan Smith [3]
[1] Department of Surgery, Arrowhead Regional Medical Center, Colton, CA; Chicago Medical School, North Chicago, IL
[2] Department of Physical Medicine and Rehabilitation, University of California, Davis, Sacramento, CA; Chicago Medical School, North Chicago, IL
[3] California University of Science and Medicine, Colton, CA
Correspondance: Sean.Hormozian@my.rfums.org
ABSTRACT
The development of cardiovascular disease is largely attributed to upstream endothelial dysfunction in the vasculature. While the exact mechanisms at the cellular level are not fully understood and always evolving, recent research has shown support for an interaction between macrophages and the sympathetic nervous system that amplifies endothelial damage in disease states. Specifically, studies have focused on neuro-immune modulation in the context of obesity and hypertension. It appears that the innate immune system responds to adrenergic stimulation through various receptors and agonists, such as norepinephrine, to increase endothelial damage and further the risk for heart disease.
INTRODUCTION
Cardiovascular disease (CVD) currently stands as the leading cause of death in the United States. While there are a multitude of risk factors that leave individuals vulnerable to heart disease, obesity and hypertension (HTN) have proven to play significant roles. The effects of obesity and HTN at a cellular level leading to endothelial dysfunction may explain their role as risk factors for heart disease (1).
Endothelial dysfunction in obesity is correlated with an increase in macrophage proinflammatory expression (2,3). In an experiment where researchers indicated macrophages with F3/80+, they demonstrated that there is an increase in this cell type in areas dense with adipose tissue in obese mice compared to lean mice (4). Conversely, the role of adrenergic stimulation has also been associated with endothelial changes in obesity. Sympathetic stimulation in the vasculature has simply been shown to be higher at baseline for obese patients when compared to non-obese patients (5). Researchers have even shown that adrenergic stimulation may have differing consequences in the vasculature when comparing obese to non-obese patients. After administration of a β2 adrenergic receptor (AR) agonist, one study shows that the augmentation index - a measure of vascular endothelial function - decreased significantly for only obese patients when compared to non-obese patients (6).
These trends are further mirrored in the pathogenesis of HTN. Using microneurography to measure efferent postganglionic muscle sympathetic nerve activity, Grassi et al. demonstrated that there is a significant and proportional climb in peripheral nerve activity when comparing normotensive individuals to individuals with severe essential HTN (7). Furthermore, studies with mice show a significant increase in the number of macrophages within the vasculature of hypertensive mice with left ventricular hypertrophy when compared to healthy mice (8). Similar results have been found by other researchers (9), and this concept was also recently reviewed by Drummond et al (10). Thus, it is well documented that the innate and sympathetic nervous system (SNS) independently contribute to changes in human vasculature.
What we aim to investigate in this review is how macrophages from the innate immune system are interacting with the SNS within the vasculature to affect the structure of the endothelial cells and contribute to the pathogenesis of upstream diseases that increase the risk for developing CVD. Macrophages populate arterial walls and recent studies have shown that immune cells share anatomical localization with peripheral neurons in the vasculature, hinting towards a potential neuro-immune interaction (11, 12). Certain macrophages have been found to express both α and β ARs, and are able to respond to various concentrations of norepinephrine (NE) (13). Many studies we will cite in this review suggest that there is a physiological connection between NE and macrophages that we are failing to consider when looking at endothelial homeostasis. While the exact mechanisms remain unclear, there is strong support for neuroimmunomodulation that may play an important role in various disease states. It is important to analyze these two systems within the vasculature and find roles in which they may communicate so that we gain a better understanding of the influence they have on one another in the development of heart disease. We will begin by introducing molecular findings on the interactions between macrophages and adrenergic stimulation and then discuss these effects on the development of obesity and HTN.
DRIVING VASCULAR CHANGES
Macrophages
Macrophages and the SNS are both known to interact with vasculature (14, 15). Independently, macrophages play a key role in driving vascular dysfunction (14). In the vessel wall, there are resident macrophages that populate the vascular layers that arise in the vessels shortly after birth as well as originate from bone marrow derived monocytes (11). Macrophage differentiation, proliferation, and survival in the vessel is regulated by macrophage colony stimulating factor (m-CSF), and when m-CSF was depleted in deoxycorticosterone acetate (DOCA)-salt hypertensive mice, there was an associated reduction in vascular remodeling, endothelial dysfunction, NADPH oxidase (NOX) activation, and vascular inflammation in the mesenteric artery (16). This reduction in vascular inflammation and NOX activation is expected, as macrophages are recruited to the vasculature during inflammation and express NOX to produce superoxide, a reactive oxidative species which helps drive endothelial dysfunction in HTN (17). In a study by Nuki et al., blood flow was augmented in mice by ligating the left common carotid artery which increased the luminal diameter of the right common carotid artery. In this augmented group, they found an increase in the number of macrophages in the vessel wall. When they depleted macrophages, there was no alteration in the luminal diameter and a reduction in vascular remodeling through matrix metalloproteinases (MMPs) (14). Extracellular matrix metabolism is regulated by MMPs and their respective inhibitors, tissue inhibitors of metalloproteinases (TIMPs). Galis et al. showed that human atherosclerotic plaques had an increased amount of immunoreactive macrophages and activated MMPs and decreased TIMPs compared to normal vessel walls (18). Other studies have shown a similar imbalance of circulating MMPs and TIMPs in patients with premature coronary atherosclerosis (19), as well as a protective effect against atherosclerosis with the loss of MMP-9 expression (20), implicating an increased level of active MMPs and decreased TIMPs in the development of atherosclerotic disease. There is also strong evidence that macrophages do decrease luminal diameter in obesity by reducing the levels of gas transmitters in the vessels (21).
Sympathetic Nervous System (SNS)
Independently, the SNS also plays a key role in driving endothelial dysfunction. It produces the catecholamine NE, which can bind α1 ARs in smooth muscle cells located in the vasculature, causing vasoconstriction (22). Vascular endothelial cells have also been found to express various subtypes of α1 ARs, regulating vasoconstriction or dilation (15). In inflammatory or diseased states, there is an increased activation of the SNS, leading to higher amounts of NE in the plasma. Under this increased activation of perivascular nerves, structural changes occur in endothelial cells along the arterial wall (23).
Anatomic Location
Given their individual interactions, it is likely that macrophages and the SNS together play a role in the upstream contributions to CVD. However, the mechanisms are complex and widespread. When plasma NE is elevated, CD14+ monocytes demonstrate increased adhesion to endothelial cells, identifying one of the initial steps in how SNS and macrophage crosstalk contributes to endothelial dysfunction leading to CVD (17). Many immune cells are known to share anatomical localization with peripheral neurons, indicating local neuro-immune interactions may have an effect on tissue homeostasis and inflammation (12). For example, certain studies show that NE released in nerve terminals contains chemoattractant properties that help guide macrophages and monocytes towards them (24).
This local neuro-immune interaction is well defined in the gut. The gastrointestinal (GI) tract is innervated by the enteric nervous system (ENS), a division of the autonomic nervous system that helps regulate the function of the GI tract. Macrophages located in the muscularis mucosa were found to preferentially express β2ARs and reside near the myenteric plexus. Upon stimulation of the β2AR with NE, these macrophages upregulated anti-inflammatory genes and became tissue protective (Figure 1) (25). The ENS and macrophage have a reciprocal relationship to maintain survival. Macrophages in the muscularis externa release bone morphogenic protein-2, a protein which acts on neurons in the gut to maintain peristalsis, and the myenteric plexus release m-CSF, a growth colony stimulating factor contributing to the survival of macrophages (26). We speculate that a similar relationship could be occurring in the vasculature, however it may lead to endothelial dysfunction instead.

Figure 1: Adrenergic regulation of immune profile in macrophages. Macrophages are known to express M0 (non-activated), M1 (pro-inflammatory), or M2 (anti-inflammatory) phenotypes; the latter two contribute different cytokines to their environment. Cytokines such as TNFα, IL-6, and IL-1 are released from M1 macrophages and promote inflammation and damage in the vasculature and tissues. Cytokines such as transforming growth factor β (TGFβ), IL-10, and IL-4 are released from M2 macrophages and act to suppress the inflammatory response and promote healing. There are different environmental factors that help macrophages polarize towards either an M1 or M2 identity, but one recently investigated driver is stimulation of their ARs. While it has been defined for a long time now that LPS and interferon γ (IFN-γ) promote the M1 phenotype and IL-4 and IL-13 promote the M2 phenotype, studies are beginning to support the notion that stimulation through the α1, α 2, and β1 ARs on macrophages promotes the M1 phenotype, while stimulation of the β2 AR promotes the M2 phenotype
CROSSTALK ON OBESITY
Obesity is a disease that is characterized by an excess amount of adipose tissue which leads to a higher amount of pro-inflammatory gene expression and a reduced expression of anti-inflammatory genes (27). In this state of inflammation, patients are at increased risk of developing complications, such as CVD. The macrophage plays a large role in the development of inflammation in obesity. In areas dense with adipose tissue there is an increase in pro-inflammatory macrophages, which were a significant source of tumor necrosis factor α (TNFα), interleukin-6 (IL-6), and inducible nitric oxide synthase, all of which are pro-inflammatory cytokines (4).
SNS overactivity is also implicated in obesity and a known driver of obesity induced HTN. In normotensive obese individuals there is marked increase in sympathetic nerve firing and this increased sympathetic output increases blood pressure and cardiac output (5). As we can see, both sympathetic overactivation and macrophages are playing a role in facilitating downstream effects in obesity, but is there an interaction between macrophages and the SNS, furthering the dysfunction?
Sympathetic neuron-associated macrophages (SAMs) in adipose tissue are known to be upregulated during obesity and can directly alter adipocyte access to NE (28). They uptake NE through the solute carrier family 6 member 2 (SLC6A2) transporter protein and possess the ability to metabolize NE through monoamine oxidase A (MAO-A). When mice were treated with SLC6A2 ablation, they demonstrated an anti-obesity effect as there was now more NE in the adipocytes, leading to increased lipolysis and weight loss through thermogenesis (27). Furthermore, it was found that MAO-A expression is regulated by the NOD-like receptor protein 3 (NLRP3) inflammasome, and when this is inhibited, there is improved lipolysis due to increased NE availability (28). When the SNS is stimulated with optogenetics to upregulate NE uptake by SAMs, these SAMs increase expression of TNFα and IL-1α, cytokines that contribute to proinflammatory state and overall endothelial dysfunction, linking to the development of CVD (27).
Obesity can be induced in mice through a high fat diet Dahl salt-sensitive (HFD Dahl SS) method, and it also serves as an excellent model in studying HTN in obese mice. In a recent study by Mui et al., the researchers looked to see if dysfunction exists in the interaction between the mesenteric arteries and the α2ARs on the superior mesenteric and celiac ganglia (SMCG) of the SNS in driving HTN in this model. They found that systolic blood pressure was raised and there was an increase in macrophage accumulation in the mesenteric artery. However, there was a decrease in monocyte chemoattractant protein-1 and TNFα compared to normal fat diet mice, both proinflammatory cytokines. Unlike the DOCA-salt hypertensive model, prejunctional α2AR dysfunction was not detected in SMCG neurons and is not a likely contributor to obesity related HTN in this model. The authors point out that although this dysfunction was not seen in the mesenteric vascular bed, it is possible the α2AR dysfunction may occur in other organs important in blood pressure regulation such as the kidneys (29). Another study using HFD Dahl SS mice reports that in males, but not females, development of HTN may be driven by a transient and mild increase in neurovascular transmission driving vasoconstriction in mesenteric arteries, but it is not likely implicated in maintenance of HTN (30). These studies show that the pathogenesis of obesity driven HTN is multifactorial and that alterations in the vascular sympathetic neurotransmission and macrophages are not entirely responsible for the development of this complication.
Leptin seems to be a key intermediate in propagating the connection between pro-inflammatory macrophages and the SNS. Adipocytes secrete the adipokine leptin, which is a hormone increased in obesity that plays numerous roles in the disease. When adipose tissue macrophages were treated with leptin, they paradoxically expressed the M2 surface markers (IL-4r) but were able to secrete proinflammatory cytokines such as TNFα, IL-6, and IL-1β (31). Hyperleptinemia is also a potent stimulator of the SNS. Carlyle et al. studied rats with increased leptin and recorded an overall increase in mean arterial pressure (MAP) and heart rate over seven days. When treated with an α AR antagonist, chronic leptin infusion did not cause an SNS-induced increase in MAP (32).
CROSSTALK ON HYPERTENSION (HTN) RELATED TO DEVELOPING CARDIOVASCULAR DISEASE (CVD)
HTN increases the risk for CVD. It has long been understood that overactivation of the SNS and the response of baroreceptors plays a significant role in this development (7), but it is also well studied that the innate immune response through the function of macrophages contributes to vessel wall thickening and hypertensive heart disease (8). Recent research has focused more on the interaction between these two drivers of HTN and atherosclerosis in the development heart disease. In a study focusing on interleukin-6 (IL-6) messenger RNA (mRNA) expression in cell cultures, a team of researchers found a time and concentration dependent rise in IL-6 from U937 resident macrophages after administration of NE. They attributed the increased production of IL-6 to an interaction between NE and macrophages involving the β adrenoreceptor-reactive oxygen species-NF-kB signal pathway (33).
IL-6 may be a strong link between the effects of adrenergic stimulation and macrophages on promoting vascular inflammation leading to heart disease. A three-year prospective case control study published in the New England Journal of Medicine looked at various inflammatory markers as a predictor of CVD in post-menopausal women with no reported underlying health conditions. They found elevated levels of IL-6 in the plasma to be strongly correlated with the risk of future cardiovascular events in this population (34). This is further supported by recent research that discovered phagocytic cells from the innate immune system, such as macrophages, synthesize and release catecholamines under inflammatory conditions. When macrophages face insult such as with lipopolysaccharide (LPS), they release NE to act in an autocrine fashion in order to promote the release of cytokines such as IL-1β and TNFα (13). These cytokines play crucial roles in vascular remodeling during inflammatory states, promoting smooth muscle migration and leading to increased likelihood of CVD secondary to HTN (Figure 2).
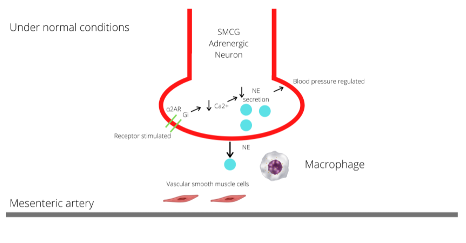
Figure 2A: SMCG of the SNS located near the mesenteric artery. Under normal conditions, the prejunctional α2 AR inhibits NE release through Gi proteins which inhibit voltage gated N-type Ca2+ channels that control NE release, this negative feedback allows regulation of sympathetic tone and increases in blood pressure (28).

Figure 2B: SMCG of the SNS located near the mesenteric artery. In DOCA-salt hypertensive mice, macrophages invade the synapse and can produce superoxide, which in turn cause prejunctional internalization of the α2 AR. When the α2 AR is internalized, this inhibits the regulation of NE production and secretion, so now NE is secreted unopposed, worsening the inflammation and vasoactive effects on the mesenteric artery. When macrophages are stimulated by α2 AR, they produce pro-inflammatory cytokines IL-1β, TNFα, and IL-6 which go on to further exacerbate the inflammatory effects and vascular remodeling. This vascular remodeling is the initial step in the development of CVD.
Macrophages and adrenergic receptors interact through free radicals, and this has been well studied in the mesenteric arteries in DOCA-salt hypertensive rats. The mesenteric arteries are major resistance arteries and large contributors to the development of HTN in this model. These arteries are innervated by the superior mesenteric and celiac ganglia of the SNS. Under normal conditions, the prejunctional α2 AR inhibits NE release from sympathetic nerves through Gi proteins which inhibit voltage gated N-type Ca2+ channels that control NE release. This negative feedback allows for regulation of sympathetic tone and increase in blood pressure (29). A study conducted by Thang et al. found that macrophage NOX produces superoxide which interacts with the α2 ARs in the vasculature to raise blood pressure. Analyzing the mesenteric artery in DOCA-salt hypertensive rats, macrophages recruited to the vasculature led to an increase in levels of superoxide in the vascular adventitia compared to rats who were not treated with mineralocorticoid-salt excess, and thus had no significant increase in their SNS activation. Superoxide acts to impair prejunctional α2 AR receptors by causing receptor internalization, which leads to an increase in production of NE due to disruption in feedback inhibition (Figure 2) (29). Therefore, macrophages that were recruited because of HTN in the DOCA-salt rats were ultimately leading to greater sympathetic activation via decreased inhibition of NE release (35). This becomes a positive feedback loop as studies also show that NE increases superoxide production through stimulation of α2 AR on peripheral blood monocytes (17). This was measured with p22phox mRNA expression and stimulated a similar physiological response as when macrophages contact a pro-inflammatory marker such as LPS. However, peripheral blood monocytes were not able to be isolated and instead the CD14 marker was obtained as an indicator of macrophages. In addition to raising blood pressure, these interactions ultimately lead to greater vascular remodeling and further increase the risk of CVD (16).
It is important to mention that in other studies with similar models, they found that adenosine is produced by the perivascular SNS in the mesenteric arteries and can bind to the adenosine 1 receptor in the periarterial nerves. This disrupts NE regulation, resulting in a greater increase and providing an alternative pathway to explain the rise of NE levels (36).
CONCLUSIONS
Potential Therapeutic Strategies
As we move forward, we must not only consider the independent interactions of macrophages and SNS in driving endothelial dysfunction, but also how their intimate interaction is contributing to a pathologic state. The general trend we have seen is that the SNS interacting with macrophages through physiological NE stimulation has supported a pro-inflammatory response leading to endothelial damage and increased likelihood of CVD (Figures 1, 2). We have also seen macrophages interacting directly with sympathetic neurons in the mesenteric artery, furthering the development of hypertensive disease (29). However, the pro-inflammatory profile of circulating monocytes and macrophages seems to be altered under β2 AR stimulation. In a study done by Galvez et al., high fat diet mice were used as a model of obesity and compared to standard diet in control lean mice. As expected, the circulating monocytes in the obese group expressed inflammatory cytokines (TNFα, IL-1, IL-6). When stimulated with β2 AR agonist terbutaline, the pro-inflammatory monocytes in obese mice shifted to an anti-inflammatory gene expression, with an increase in anti-inflammatory cytokines (IL-10, IL-4, and IL-5) (37). Another laboratory found results supporting these findings after treating Zucker diabetic fatty rats with a β2 AR agonist for 12 weeks. They concluded that this mediated the inhibition of inflammatory cytokine production and lowered monocyte activation, speculating that the β2 AR agonists may have protective effects against diabetic cardiovascular complications (38). These revelations provide therapeutic thought to work upstream and prevent the development and progression of CVD in obese individuals.
Limitations and Future Directions
We still face many limitations and topics that need to be further studied. As previously noted, the mechanisms of neuro-immune crosstalk in developing and maintaining HTN in high-fat diet mice is different from the DOCA-salt model at the mesenteric arteries (29). This notion is important as it furthers the evidence that the molecular mechanisms contributing to the pathogenesis of HTN and CVD is quite complex, and there is still a lot of work to be done under the topic of neuro-immune interaction.
Furthermore, the SNS is multi-level depending on the initial stimulus and power of inflammation. The effects of the SNS with macrophages in perivascular adipose tissue can vary depending on the initial driver of stimulation, making potential pharmacological treatment complicated. For instance, studies have shown that a healthy increase in the SNS, such as exercise, can promote the M2 phenotype for macrophages. Alternatively, a pathological increase, such as a high fat diet, can promote the M1 phenotype for macrophages (39). Thus, the neuro-immune crosstalk may differ for individuals depending on comorbidities and the effects on vascular disease may vary not only by the quality of their interaction, but also with proper timing.
We have highlighted various neuro-immune interactions that may contribute to upstream risk factors for CVD, but there are still more factors that need to be studied. For example, recent studies hint at the possibility of hydrogen sulfide acting as a liaison between macrophages and the SNS in vascular homeostasis and atherosclerosis (21, 40). Ultimately, with greater research in this subject we may be able to target and treat the upstream vascular pathology that aids in the development of CVD more effectively.
DISCLOSURES
Funding: Not applicable.
Conflicts of interest: None.
Availability of data and materials: Upon request.
Code availability: Not applicable.
Authors’ contributions: Authors listed in the manuscript have contributed per submission guidelines and standards for authorship.
Ethics approval: Not applicable.
Consent to participate: Not applicable.
REFERENCES
1. Aggoun, Y. et al. Impaired endothelial and smooth muscle functions and arterial stiffness appear before puberty in obese children and are associated with elevated ambulatory blood pressure. Eur. Heart J. 29, 792–799 (2008).
2. Sledzinski, T., Sledzinski, M., Smolenski, R. T. & Swierczynski, J. Increased serum nitric oxide concentration after bariatric surgery—a potential mechanism for cardiovascular benefit. Obes. Surg. 20, 204–210 (2010).
3. Apovian, C. M. et al. Adipose macrophage infiltration is associated with insulin resistance and vascular endothelial dysfunction in obese subjects. Arterioscler. Thromb. Vasc. Biol. 28, 1654–1659 (2008).
4. Weisberg, S. P. et al. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 (2003).
5. Grassi, G. et al. Sympathetic activation in obese normotensive subjects. HTN 25, 560–563 (1995).
6. Suh, H.-S. et al. Vascular endothelial dysfunction tested by blunted response to endothelium-dependent vasodilation by salbutamol and its related factors in uncomplicated pre-menopausal obese women. Int. J. Obes. 29, 217–222 (2005).
7. Grassi, G., Cattaneo, B. M., Seravalle, G., Lanfranchi, A. & Mancia, G. Baroreflex control of sympathetic nerve activity in essential and secondary HTN. HTN 31, 68–72 (1998).
8. Kain, D. et al. Macrophages dictate the progression and manifestation of hypertensive heart disease. Int. J. Cardiol. 203, 381–395 (2016).
9. Kai H., Kudo H., Takayama N., Yasuoka S., Kajimoto H., Imaizumi T. Large blood pressure variability and hypertensive cardiac remodeling--role of cardiac inflammation. Circ J. 2009;73(12):2198-2203. doi:10.1253/circj.cj-09-0741
10. Drummond, G. R., Vinh, A., Guzik, T. J. & Sobey, C. G. Immune mechanisms of HTN. Nat. Rev. Immunol. 19, 517–532 (2019).
11. Ensan, S. et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat. Immunol. 17, 159–168 (2016).
12. Kabata, H. & Artis, D. Neuro-immune crosstalk and allergic inflammation. J. Clin. Invest. 130, 1475–1482 (2019).
13. Spengler, R. N., Chensue, S. W., Giacherio, D. A., Blenk, N. & Kunkel, S. L. Endogenous NE regulates tumor necrosis factor-alpha production from macrophages in vitro. J. Immunol. 152, 3024–3031 (1994).
14. Nuki, Y. et al. Roles of macrophages in flow-induced outward vascular remodeling. J. Cereb. Blood Flow Metab. 29, 495–503 (2009).
15. Filippi, S. et al. α1D-Adrenoceptors Cause Endothelium-Dependent Vasodilatation in the Rat Mesenteric Vascular Bed. J. Pharmacol. Exp. Ther. 296, 869–875 (2001).
16. Ko, E. A. et al. Resistance artery remodeling in deoxycorticosterone acetate-salt HTN is dependent on vascular inflammation: evidence from m-CSF-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 292, H1789–95 (2007).
17. Deo, S. H., Jenkins, N. T., Padilla, J., Parrish, A. R. & Fadel, P. J. NE increases NADPH oxidase-derived superoxide in human peripheral blood mononuclear cells via α-adrenergic receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R1124–32 (2013).
18. Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94(6):2493-2503. doi:10.1172/JCI117619
19. Noji Y, Kajinami K, Kawashiri MA, et al. Circulating matrix metalloproteinases and their inhibitors in premature coronary atherosclerosis. Clin Chem Lab Med. 2001;39(5):380-384. doi:10.1515/CCLM.2001.060
20. Luttun A, Lutgens E, Manderveld A, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109(11):1408-1414. doi:10.1161/01.CIR.0000121728.14930.DE
21. Candela, J., Wang, R. & White, C. Microvascular Endothelial Dysfunction in Obesity Is Driven by Macrophage-Dependent Hydrogen Sulfide Depletion. Arterioscler. Thromb. Vasc. Biol. 37, 889–899 (2017).
22. Cipolla, M. J., Harker, C. T. & Porter, J. M. Endothelial function and adrenergic reactivity in human type-II diabetic resistance arteries. J. Vasc. Surg. 23, 940–949 (1996).
23. Loesch, A., Maynard, K. I. & Burnstock, G. Calcitonin gene-related peptide- and neuropeptide Y-like immunoreactivity in endothelial cells after long-term stimulation of perivascular nerves. Neuroscience 48, 723–726 (1992).
24. Straub, R. H. et al. Neurotransmitters of the sympathetic nerve terminal are powerful chemoattractants for monocytes. J. Leukoc. Biol. 67, 553–558 (2000).
25. Gabanyi, I. et al. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 164, 378–391 (2016).
26. Stakenborg, N., Viola, M. F. & Boeckxstaens, G. E. Intestinal neuro-immune interactions: focus on macrophages, mast cells and innate lymphoid cells. Curr. Opin. Neurobiol. 62, 68–75 (2019).
27. Pirzgalska, R. M. et al. Sympathetic neuron–associated macrophages contribute to obesity by importing and metabolizing NE. Nat. Med. 23, 1309–1318 (2017).
28. Camell, C. D. et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123 (2017).
29. Mui, R. K., Fernandes, R. N., Garver, H. G., Van Rooijen, N. & Galligan, J. J. Macrophage-dependent impairment of α2-adrenergic autoreceptor inhibition of Ca2+ channels in sympathetic neurons from DOCA-salt but not high-fat diet-induced hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 314, H863–H877 (2018).
30. Alula, K. M. et al. Effects of high-fat diet on sympathetic neurotransmission in mesenteric arteries from Dahl salt-sensitive rat. Auton. Neurosci. 222, 102599 (2019).
31. Acedo, S. C., Gambero, S., Cunha, F. G. P., Lorand-Metze, I. & Gambero, A. Participation of leptin in the determination of the macrophage phenotype: an additional role in adipocyte and macrophage crosstalk. In Vitro Cell. Dev. Biol. Anim. 49, 473–478 (2013).
32. Carlyle Megan, Jones Oscar B., Kuo Jay J. & Hall John E. Chronic Cardiovascular and Renal Actions of Leptin. HTN 39, 496–501 (2002).
33. Li, M., Yao, W., Li, S. & Xi, J. NE induces the expression of interleukin-6 via β-adrenoreceptor-NAD(P)H oxidase system -NF-κB dependent signal pathway in U937 macrophages. Biochem. Biophys. Res. Commun. 460, 1029–1034 (2015).
34. Ridker, P. M., Hennekens, C. H., Buring, J. E. & Rifai, N. C-reactive protein and other markers of inflammation in the prediction of CVD in women. N. Engl. J. Med. 342, 836–843 (2000).
35. Thang, L. V. et al. Macrophage depletion lowers blood pressure and restores sympathetic nerve α2-adrenergic receptor function in mesenteric arteries of DOCA-salt hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 309, H1186–97 (2015).
36. Sangsiri, S., Dong, H., Swain, G. M., Galligan, J. J. & Xu, H. Impaired function of prejunctional adenosine A1 receptors expressed by perivascular sympathetic nerves in DOCA-salt hypertensive rats. J. Pharmacol. Exp. Ther. 345, 32–40 (2013).
37. Gálvez, I., Martín-Cordero, L., Hinchado, M. D., Álvarez-Barrientos, A. & Ortega, E. Anti-inflammatory effect of β2 adrenergic stimulation on circulating monocytes with a pro-inflammatory state in high-fat diet-induced obesity. Brain Behav. Immun. 80, 564–572 (2019).
38. Noh, H. et al. Beta 2-adrenergic receptor agonists are novel regulators of macrophage activation in diabetic renal and cardiovascular complications. Kidney Int. 92, 101–113 (2017).
39. Saxton, S. N., Withers, S. B. & Heagerty, A. M. Emerging Roles of Sympathetic Nerves and Inflammation in Perivascular Adipose Tissue. Cardiovasc. Drugs Ther. 33, 245–259 (2019).
40. Kang, S. C., Sohn, E.-H. & Lee, S. R. Hydrogen Sulfide as a Potential Alternative for the Treatment of Myocardial Fibrosis.Oxid. Med. Cell. Longev.2020, 4105382 (2020).